Die aufwendige Suche nach Fehlern in der Basenfolge
Freiburg, 04.10.2019
Foto: Jürgen Gocke
Heute sind circa 6.000 bis 8.000 seltene Erkrankungen bekannt. Viele von ihnen werden durch einen Gendefekt verursacht. Als selten gilt eine Krankheit, wenn nicht mehr als eine unter 2.000 Menschen betroffen ist. Doch auch wenn jede einzelne dieser Erkrankungen nur äußerst selten diagnostiziert wird, so sind insgesamt dennoch etwa fünf bis sieben Prozent der Bevölkerung – in Deutschland knapp vier Millionen Menschen, in Europa etwa 30 Millionen – von seltenen Erkrankungen betroffen. Die Konsequenz: Die überregionale Verteilung der wenige Fälle macht Studien sehr aufwendig, Diagnosen können oft nur mit großer Verzögerung gestellt werden, und Expertinnen und Experten, die zu seltenen Erkrankungen und Therapiemöglichkeiten forschen, sind rar. Eine von ihnen ist Dr. Miriam Schmidts. Seit 2015 arbeitet sie im Team der Sektion Pädiatrische Genetik am Zentrum für Kinder- und Jugendmedizin des Universitätsklinikums Freiburg.
 Im Labor versucht Miriam Schmidts herauszufinden, welche genetischen Defekte eine seltene Erkrankung auslösen. Foto: Jürgen Gocke
Im Labor versucht Miriam Schmidts herauszufinden, welche genetischen Defekte eine seltene Erkrankung auslösen. Foto: Jürgen Gocke
Miriam Schmidts kommt gerade von einem Beratungsgespräch mit den Eltern eines jungen Patienten, der an einer genetisch bedingten Ziliopathie leidet. Jetzt nimmt sie sich kurz Zeit, um über ihre Forschung zu berichten, und klappt ihren Laptop auf. Die bunten Animationsfilmchen, die über den Bildschirm flackern, wirken wie für die Kinder gemacht, denen sie ihre volle Aufmerksamkeit als Ärztin und ihr ganzes Interesse als Forscherin widmet. Hunderte kleine Härchen auf der Oberfläche von Zellen bewegen sich da synchron im Takt wie eine Cheerleading-Gruppe bei ihrer Performance. In sanften Wellen tanzen auf ihren Spitzen zwei dicke Fladen in Grüngelb und bewegen sich dabei langsam wie auf einem Fließband aus dem Bild.
„So soll es sein”, sagt Schmidts. „Wenn die Zilien richtig arbeiten, transportieren sie den Schleim aus der Lunge. Wenn nicht, kommt es zum Stau.” Wie im Film nebenan. Dort wackelt das Grüngelb orientierungslos auf der Stelle, weil die Härchen sich weder darüber einig sind, in welche Richtung sie sich bewegen, noch, ob sie überhaupt etwas tun sollen. Steigt der Schleim jedoch nicht auf, kann er nicht ausgehustet werden. So setzt sich die Lunge langsam zu, Bakterien sammeln sich an und es kommt zu Infektionen – wie bei der Mukoviszidose oft mit nachhaltigen Schäden.
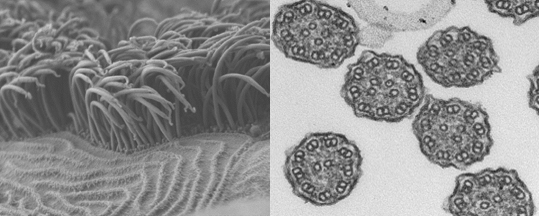 Bewegliche Zilien unter dem Elektronenmikroskop – ganz (links) und im Querschnitt: Die Aufgabe der feinen Härchen besteht darin, Schleim aus der Lunge zu transportieren.
Bewegliche Zilien unter dem Elektronenmikroskop – ganz (links) und im Querschnitt: Die Aufgabe der feinen Härchen besteht darin, Schleim aus der Lunge zu transportieren.
Quelle: Miriam Schmidts
Folge verschiedener Gendefekte
Diese Dysfunktion der Zilien ist Folge verschiedener Gendefekte, sagt Schmidts. Die kleinen Härchen, die – je nach Typ und Körperregion – mal einzeln, mal in Büscheln an jeder Zelle wachsen, wurden um das Jahr 1800 erstmals unter dem Mikroskop entdeckt. Zunächst glaubte man, sie hätten keine Funktion – bis man herausfand, dass die beweglichen unter ihnen dafür zuständig sind, organisches Material von A nach B zu transportieren: Sekret aus den Atemwegen, die Eizelle zur Gebärmutter, die Spermien in den Eileiter. Nicht bewegliche Zilien hingegen arbeiten wie Antennen auf der Zelloberfläche und sind für die Verarbeitung für verschiedene Signalwege der Zelle wichtig. Dementsprechend führen Funktionsstörungen dieser Härchen oft zu Entwicklungsstörungen verschiedener Organe wie zum Beispiel der Nieren und des Gehirns oder auch des Skelettsystems. Evolutionär gesehen stammen Zilien von Flagellen ab, wie sie auch bei Algen oder Bakterien zu finden sind.
Nach ihrer Promotion 2005 bei dem Freiburger Nephrologen Prof. Dr. Gerd Walz kam Schmidts als Assistenzärztin an die Kinderklinik des Universitätsklinikums Freiburg und arbeitete an einem Forschungsprojekt über bewegliche Zilien mit, das Prof. Dr. Heymut Omran, heute Direktor der Klinik für Kinder- und Jugendmedizin am Universitätsklinikum Münster, betreute. Anschließend ging sie nach London/England und forschte dort am University College mehrere Jahre lang über unbewegliche Zilien, die vermutlich wie Signalempfänger der Zellen funktionieren. Auch hier sind genetische Defekte verantwortlich für seltene Erkrankungen. Kinder kommen dann oft mit Hirnfehlbildungen auf die Welt – oder mit mehr als fünf Fingern pro Hand, weil bestimmte Signalwege, die für die Ausbildung der Finger notwendig sind, gestört sind.
Bis heute forscht Schmidts vor allem in diesem Bereich, insbesondere über Skelettdysplasien wie das seltene Ellis-van-Creveld-Syndrom. Bei diesen vorgeburtlichen Anomalien ist das Wachstum der Rippen gestört. Die Kinder werden mit viel zu kleinem Brustkorb geboren, können nicht richtig atmen, sterben oft bis zum zweiten Lebensjahr, ohne dass sich das medizinisch verhindern ließe. Überleben sie, bilden sie Zysten an den Nieren aus, die zu Nierenversagen führen, oder sie erblinden, weil die Fotorezeptoren in den Augen ihre Funktion aufgeben.
 Seit 2015 forscht Miriam Schmidts am Zentrum für Kinder- und Jugendmedizin des Universitätsklinikums Freiburg. Foto: Britt Schilling/Universitätsklinikum Freiburg
Seit 2015 forscht Miriam Schmidts am Zentrum für Kinder- und Jugendmedizin des Universitätsklinikums Freiburg. Foto: Britt Schilling/Universitätsklinikum Freiburg
Erkrankungsmodelle aus gesunden Zellen
„Im Labor versuchen wir herauszufinden, was genau der Gendefekt ist, der dieses Krankheitsbild erzeugt”, sagt Schmidts. „Dafür suchen wir mit den neuesten Sequenzierungstechnologien zunächst nach dem defekten Gen, das die Krankheit verursacht.“ Diese Suche sei heute deutlich einfacher als noch vor ein paar Jahren, da die entsprechenden Proben mit Gendatenbanken abgeglichen werden können, in denen die Basenfolgen von Zehntausenden gesunder Menschen gespeichert sind. „Was uns interessiert, ist: Wie verursacht das Gen den Defekt im Körper? Und warum bekommt der Körper all diese Probleme – von der Hirnfehlbildung bis zur Erblindung –, wenn dieses eine Gen fehlt?” Dafür erstellt Schmidts nach Vorlage von Zellproben der Patientinnen und Patienten Erkrankungsmodelle aus gesunden Zellen, bei denen mit der CRISPR/CAS-Methode dann jener Gendefekt eingebaut wird, den auch die Patientin oder der Patient hat.
Dass diese Methode in Zukunft auch zur Heilung oder zur Korrektur von Gendefekten genutzt werden könnte, indem man auf diese Weise Fehler in der Basenfolge bereinigt, hält Schmidts zwar theoretisch für möglich, aber aus ethischen und medizinischen Gründen für hochproblematisch. „Es ginge hier immerhin um eine Veränderung des menschlichen Erbguts, deren Nebenwirkungen und Langzeitfolgen wir in keiner Weise abschätzen könnten.” Eines der zentralen Ziele ihrer Grundlagenforschung ist deshalb die Entwicklung neuer Therapien für diese seltenen Erkrankungen – und das nicht nur im Bereich der Gentherapie. „Wenn wir etwa bestimmte Signalwege in Zellen mit pharmakologischen Mitteln manipulieren könnten, wären deutliche Verbesserungen für die Patienten denkbar.“ Anhand von Algen, deren Flagellen die gleiche Zilienstruktur wie beim Menschen aufweisen, testen Schmidts und ihr Team zudem viele bereits zugelassene Substanzen auf möglicherweise noch unbekannte Wirkung auf die Zilien.
Kontakt mit Patienten und deren Familien
Wichtig ist Miriam Schmidts, die selbst junge Mutter ist, neben der Forschung auch der Kontakt mit den jungen Patienten und den betroffenen Familien: „Als Ärztin möchte ich, dass die Eltern verstehen, warum ihr Kind ist, wie es ist. Auch wenn die Krankheiten oft nicht heilbar sind und nicht selten tödlich verlaufen, ist das wichtig, da gerade bei genetisch bedingten Krankheiten der psychologische Druck für die Eltern enorm ist. Viele fragen sich, was sie falsch gemacht haben. Da hilft es, ihnen sagen zu können, dass die Erkrankung nicht zu vermeiden gewesen wäre.” Und auch, ihnen zu helfen, das Risiko realistisch einzuschätzen, ob ein weiteres Kind erneut von der gleichen Krankheit betroffen sein könnte.
Da klingelt das Telefon. Miriam Schmidts klappt den Laptop zu und entschuldigt sich. Da müsse sie jetzt ran. Die Patienten und ihre Angehörigen haben zu jeder Zeit ihr offenes Ohr.
Dietrich Roeschmann